
Looking for a valid and free Office 2021 product key? Wondering how to activate Microsoft Office 2021? Your search ends here! We have collected tons of free product key for Microsoft Office 2021 and today we will share them with you! Besides, there are also some insider tips to activate Office 2021 without product key that you absolutely can't miss! Without further delay, let's get started!
- Part 1. How To Activate Microsoft Office 2021 With Product Key?
- [Tutorial] Activate MS Office 2021 With Product Key
- [Collection] FREE List Of Office 2021 Product Key
- Part 2. How To Activate Office 2021 Without Product Key?
- Part 3. How To Crack Encrypted Excel/Word/PowerPoint Without Password?

Part 1. How To Activate Microsoft Office 2021 With Product Key?
Microsoft Office 2021 is a version of the Microsoft Office application for Windows and macOS operating systems. As you know, Microsoft office has a 30-day trial period, after which you will need a Microsoft office product key to activate, otherwise it will not work. It means that you have to pay for it. That's why many people are looking for free Office 2021 product key or activation code serial number. So, how to find free and valid Microsoft Office 2021 product key? And, how to activate Office 2021 with product key?
[Tutorial] Activate MS Office 2021 With Product Key
Step 1: Download Microsoft Office 2021 from Microsoft official website.
Step 2: Open one of the Office apps like Excel, Word. Click the "Sign in" button.
Step 3: Select "Activate Product".

Step 4: Click "I have a product key". Then enter the Microsoft Office 2021 activation key. Then click "Activate Office".

Step 5: After the activation, you can see a "Product Activated" message along with the Office edition on the Account page.
[Collection] FREE List Of Office 2021 Product Key
Here is the list of the latest Microsoft Office 2021 product key. You can select any one to activate Microsoft Office 2021.
Product Key for Microsoft Office 2021
5RXE4S-SX5DCR-TFV7BG-8HUN9IJ-MN8H
MNH8-UT6D5R-ESXDR-C6TFVGB-YHUNIJ
ZAQ3W-S4XED5-CRF6TV-B8HYN-UJ9IHU
V6BG8H-UNIJM9-8NHBG-TFV6D-5RE4D5
XCRTFV-BGHUNIJ-9M09N-HBGY7-TFVDC
8BYV7T-6CD5RX-E4SZEX-TFV68N-HUIJM
K0OMIJ-98HUBVY-7TDC5-RESX4-SXTFV6
8NHUIJ9-M0KJ9N-H8UTFV6-D5RXE-4STF
UTFV6D-5RESX4-X5DC6TF-V7B8H-UN9IJ
CDMPW-BKNR6-MBMWR-RTYBD-DV9JK
KDNJ9-G2MPB-HWJB4-DC6C2-DDCWD
NMMKJ-6RK4F-KMJVX-8D9MJ–6MWKP
6NPT7-HJM3B-PWC77-MHT46-QYMFX
DJY4M-6NY4K-K3PWP-YYB2M-FGGWK
T4NMF-GGX86-XGFXM-93CGC-GRBFX
AZXCD-BGHTR-CVFDR-JHYTE-XCDSW
MPN8J-8MYK2-9W92H-GTKJW-VH9JK
K7KQK-N3CHM-R2KHH-4VJ7F-TVPB9
NQQCC-86FK4-YPRCD-9CHR2-8XFY9
K9HYV-NKVKY-KBD3T-VFB2M-39Q4X
C8N7V-BFT94-KXMP3-XTGM8-TRBFX
45XCD-87BVG-54DFR-67VBG-89NBH
AZS45-XCD76-BVG98-GHY90-CFD89
GTJNT-9Q978-QJCJP-D672G-GMXJK
W7JGX-HKN4B-G8RPJ-C99CF-37C8K
TKV9N-VC8D2-XJBFC-9B7B4-R3P8K
PIU4YT-HG7FD-EW6QP-HGFD8QA
KJH3GF-NBV4GT-GFX4BN-JH8BVC
BVC1FD-JHGV2C-JHB8VC-KJH9GF
ZXC2V-JHGF7E-ASDF5G-LKJH3GK
Part 2. How To Activate Office 2021 Without Product Key?
If you are surprisingly so unfortunate that these Office 2021 product keys above don't work for you, then this method below will allow you to activate MS Office 2021 without a product key.
[Tutorial] Office 2021 Activation Without Product Key
Step 1: After installing MS Office 2021, run Command Prompt (CMD) as Administrator.
Step 2:
Paste and enter below code to specify the Office 2021 install location.
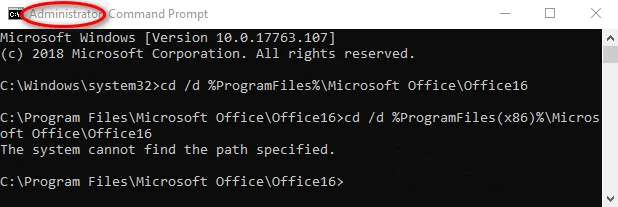
cd /d %ProgramFiles%\Microsoft Office\Office16
cd /d %ProgramFiles(x86)%\Microsoft Office\Office16
Step 3:
Use below code to covert MS Office 2021 retail license to volume license.
for /f %x in ('dir /b ..\root\Licenses16\ProPlus2021VL_KMS*.xrm-ms') do cscript ospp.vbs /inslic:"..\root\Licenses16\%x"
Step 4: Make sure your PC is connected to the internet. Then enter following command.
cscript ospp.vbs /inpkey:FXYTK-NJJ8C-GB6DW-3DYQT-6F7TH
cscript ospp.vbs /unpkey:BTDRB >nul
cscript ospp.vbs /unpkey:KHGM9 >nul
cscript ospp.vbs /unpkey:CPQVG >nul
cscript ospp.vbs /sethst:kms8.msguide.com
cscript ospp.vbs /setprt:1688
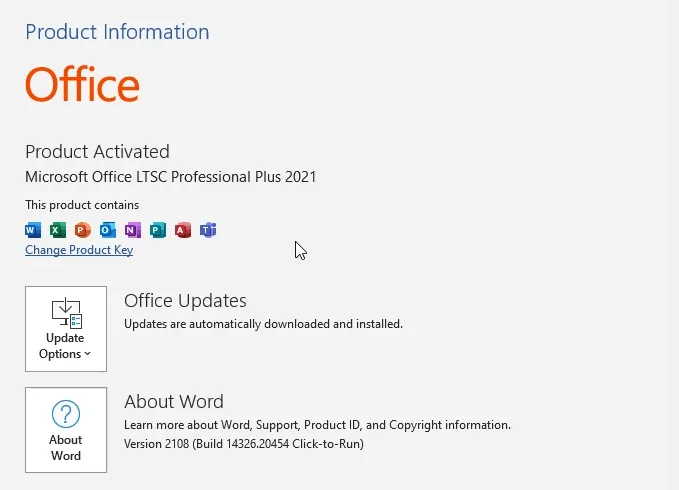
cscript ospp.vbs /act
Done! Your office 2021 is activated!
Part 3. How To Crack Encrypted Excel/Word/PowerPoint Without Password?
Microsoft Office has a very useful feature - adding passwords to Excel, Word, PowerPoint files to encrypt them and ensure that they are not freely accessed by others, protecting file privacy and data security. Yet we have to admit how common it is to forget the password or lose it! So how to unlock Excel, Word, PowerPoint without password? You can't miss out this Office file password recovery tool - WooTechy iCrowbar.
This software is designed for Microsoft Office files. It can not only recover open password of Excel, Word, PowerPoint and even PDF, ZIP, RAR, so that you can access encrypted files even if you forget the password, but also remove editing restriction on Office files, allowing you to edit, modify and copy read-only files.
WooTechy iCrowbar - Best Office File Password Cracker
8,000,000+
Downloads
- Password Recovery: Quickly recover passwords of Excel/Word/PowerPoint/PDF/ZIP/RAR to open locked files
- Restriction Removal: 1 click to remove editing restrictions on Excel/Word/PowerPoint/PDF to modify read-only files
- Multi-Language Support: 10+ multilingual passwords are supported, including English, Spanish, French, German, Chinese, etc
- AI Technology: Built-in 4 advanced AI attack modes for high success rate of file unlocking
- Unlimited Usage: No limit on the number of times to use, unlock files as many as you want
- High Security: No file data will be damaged or lost, and no file content will be affected
- Easy To Use: Intuitive interface, unlock files in simple steps, friendly to non-technical people
- 30-Day Money Back Guarantee: Instant refund within 30 days if the file cannot be unlocked
-
Free Trial
Safe Download
Full Version
Safe Payment
How To Crack Excel/Word/PowerPoint Without Password?
Step 1. Download and install iCrowbar on computer. Launch it and choose the file to crack.

Step 2. Select a recovery method and click Recover to start the password recovery. (P.S: If you are not sure which model to choose, you can check the details on the official website)

Step 3. The recovered password will be displayed on the interface, now enter or copy the password to open your locked file!

Conclusion
Well, above is our collection of Microsoft Office 2021 product key and step by step tutorial on MS Office 2021 activation, you can activate your Office 2021 now! Click to get more about Microsoft 365 product key, Office 2019 product key, Office 2016 product key, Office 2013 product key. We will update the free product keys and the latest activation methods for each version of Microsoft Office regularly. Hope it will be helpful to you!
It is worth re-emphasizing that we recommend every Microsoft Office user to use this file password recovery tool iCrowbar, which can help you unlock office files quickly and also remove editing restrictions from office file with one click, allowing you to save time and improve workplace office efficiency! Just try it for free today!
WooTechy iCrowbar
Say Goodbye to Password of ZIP / RAR / PDF / Excel / Word / PowerPoint!